Parse
While {MIPTools} provides several .csv files that can be analyzed, parsing such files is difficult because of their non-rectangular structure. As such, attempting to read these files with default parameters fails.
#> # A tibble: 10 × 5
#> X1 X2 X3 X4 X5
#> <chr> <chr> <chr> <chr> <chr>
#> 1 Gene ID PF3D7_0106300 PF3D7_0106300 PF3D7_0106300 PF3D7_01063…
#> 2 Gene atp6 atp6 atp6 atp6
#> 3 Mutation Name atp6-Ala623Glu atp6-Glu431Lys atp6-Gly639Asp atp6-Ser466…
#> 4 ExonicFunc missense_variant missense_variant missense_variant missense_va…
#> 5 AA Change Ala623Glu Glu431Lys Gly639Asp Ser466Asn
#> 6 Targeted Yes Yes No No
#> 7 D10-JJJ-23 608.0 699.0 608.0 237.0
#> 8 D10-JJJ-43 20.0 30.0 20.0 0.0
#> 9 D10-JJJ-55 158.0 242.0 158.0 61.0
#> 10 D10-JJJ-5 2.0 9.0 2.0 1.0As visible, there are six rows of metadata, which specify the gene ID, gene, mutation name, etc. The remaining rows contain the data we are actually interested in: the rows contain the samples and the columns contain the positions we are interested in. The difficult part of reading in these files is that the metadata must be extracted and treated differently from the data itself.
miplicorn, therefore, provides a family of functions: read_tbl_*() which quickly read in such non-rectangular files. The functions generate a tibble where each row represents a sample and a position. Thus, there will be multiple entries for each unique sample.
cov_file <- miplicorn::miplicorn_example("coverage_AA_table.csv")
data <- miplicorn::read_tbl_coverage(cov_file)
data
#> # A tibble: 6,344 × 8
#> sample gene_id gene mutation_name exonic_func aa_change targeted coverage
#> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <dbl>
#> 1 D10-JJJ-… PF3D7_… atp6 atp6-Ala623G… missense_v… Ala623Glu Yes 608
#> 2 D10-JJJ-… PF3D7_… atp6 atp6-Ala623G… missense_v… Ala623Glu Yes 20
#> 3 D10-JJJ-… PF3D7_… atp6 atp6-Ala623G… missense_v… Ala623Glu Yes 158
#> 4 D10-JJJ-5 PF3D7_… atp6 atp6-Ala623G… missense_v… Ala623Glu Yes 2
#> 5 D10-JJJ-… PF3D7_… atp6 atp6-Ala623G… missense_v… Ala623Glu Yes 1
#> 6 D10-JJJ-… PF3D7_… atp6 atp6-Ala623G… missense_v… Ala623Glu Yes 129
#> 7 D10-JJJ-… PF3D7_… atp6 atp6-Ala623G… missense_v… Ala623Glu Yes 0
#> 8 D10-JJJ-… PF3D7_… atp6 atp6-Ala623G… missense_v… Ala623Glu Yes 0
#> 9 D10-JJJ-… PF3D7_… atp6 atp6-Ala623G… missense_v… Ala623Glu Yes 90
#> 10 D10-JJJ-… PF3D7_… atp6 atp6-Ala623G… missense_v… Ala623Glu Yes 175
#> # … with 6,334 more rowsManipulate
Amino acids
In some the user may want to convert amino acid abbreviations from the three to one letters abbreviation, or vice versa for easier interpretation of data.
data %>%
dplyr::mutate(aa_change = convert_three(aa_change))
#> # A tibble: 6,344 × 8
#> sample gene_id gene mutation_name exonic_func aa_change targeted coverage
#> <chr> <chr> <chr> <chr> <chr> <chr> <chr> <dbl>
#> 1 D10-JJJ-… PF3D7_… atp6 atp6-Ala623G… missense_v… A623E Yes 608
#> 2 D10-JJJ-… PF3D7_… atp6 atp6-Ala623G… missense_v… A623E Yes 20
#> 3 D10-JJJ-… PF3D7_… atp6 atp6-Ala623G… missense_v… A623E Yes 158
#> 4 D10-JJJ-5 PF3D7_… atp6 atp6-Ala623G… missense_v… A623E Yes 2
#> 5 D10-JJJ-… PF3D7_… atp6 atp6-Ala623G… missense_v… A623E Yes 1
#> 6 D10-JJJ-… PF3D7_… atp6 atp6-Ala623G… missense_v… A623E Yes 129
#> 7 D10-JJJ-… PF3D7_… atp6 atp6-Ala623G… missense_v… A623E Yes 0
#> 8 D10-JJJ-… PF3D7_… atp6 atp6-Ala623G… missense_v… A623E Yes 0
#> 9 D10-JJJ-… PF3D7_… atp6 atp6-Ala623G… missense_v… A623E Yes 90
#> 10 D10-JJJ-… PF3D7_… atp6 atp6-Ala623G… missense_v… A623E Yes 175
#> # … with 6,334 more rowsSort
In plotting data, it is useful to be able to control the order in which data appears. While dplyr::arrange() provides the functionality to sort numeric or character data, it lacks the ability to naturally sort alphanumeric vectors, vectors containing both letters and numerics. Furthermore, the ordering of data is not kept when fed into plotting functions. arrange_natural() attempts to address these limitations.
arrange_natural(data, sample, gene)
#> # A tibble: 6,344 × 8
#> sample gene_id gene mutation_name exonic_func aa_change targeted coverage
#> <fct> <chr> <fct> <chr> <chr> <chr> <chr> <dbl>
#> 1 D10-JJJ-1 PF3D7_… atp6 atp6-Ala623G… missense_v… Ala623Glu Yes 10
#> 2 D10-JJJ-1 PF3D7_… atp6 atp6-Glu431L… missense_v… Glu431Lys Yes 5
#> 3 D10-JJJ-1 PF3D7_… atp6 atp6-Gly639A… missense_v… Gly639Asp No 10
#> 4 D10-JJJ-1 PF3D7_… atp6 atp6-Ser466A… missense_v… Ser466Asn No 2
#> 5 D10-JJJ-1 PF3D7_… atp6 atp6-Ser769A… missense_v… Ser769Asn Yes 1
#> 6 D10-JJJ-1 PF3D7_… crt crt-Ala220Ser missense_v… Ala220Ser Yes 2
#> 7 D10-JJJ-1 PF3D7_… crt crt-Asn326Asp missense_v… Asn326Asp No 2
#> 8 D10-JJJ-1 PF3D7_… crt crt-Asn326Ser missense_v… Asn326Ser Yes 2
#> 9 D10-JJJ-1 PF3D7_… crt crt-Asn75Glu missense_v… Asn75Glu Yes 0
#> 10 D10-JJJ-1 PF3D7_… crt crt-Cys72Ser missense_v… Cys72Ser Yes 0
#> # … with 6,334 more rowsVisualize
There are an almost limitless different ways to visualize a single set of data. While miplicorn cannot address every method, it aims to simplify the creation of key figures.
Chromosome map
There are two built in ways to create chromosome maps, each with its own set of strengths and weaknesses. You can either make an interactive map or a more detailed karyoplot.
colours <- c("#006A8EFF", "#A8A6A7FF", "#B1283AFF")
map <- plot_chromoMap(genome_Pf3D7, probes, colours = colours)
# Used to embed into html
widgetframe::frameableWidget(map)
plot_karyoploteR(genome_Pf3D7, probes, colours = colours)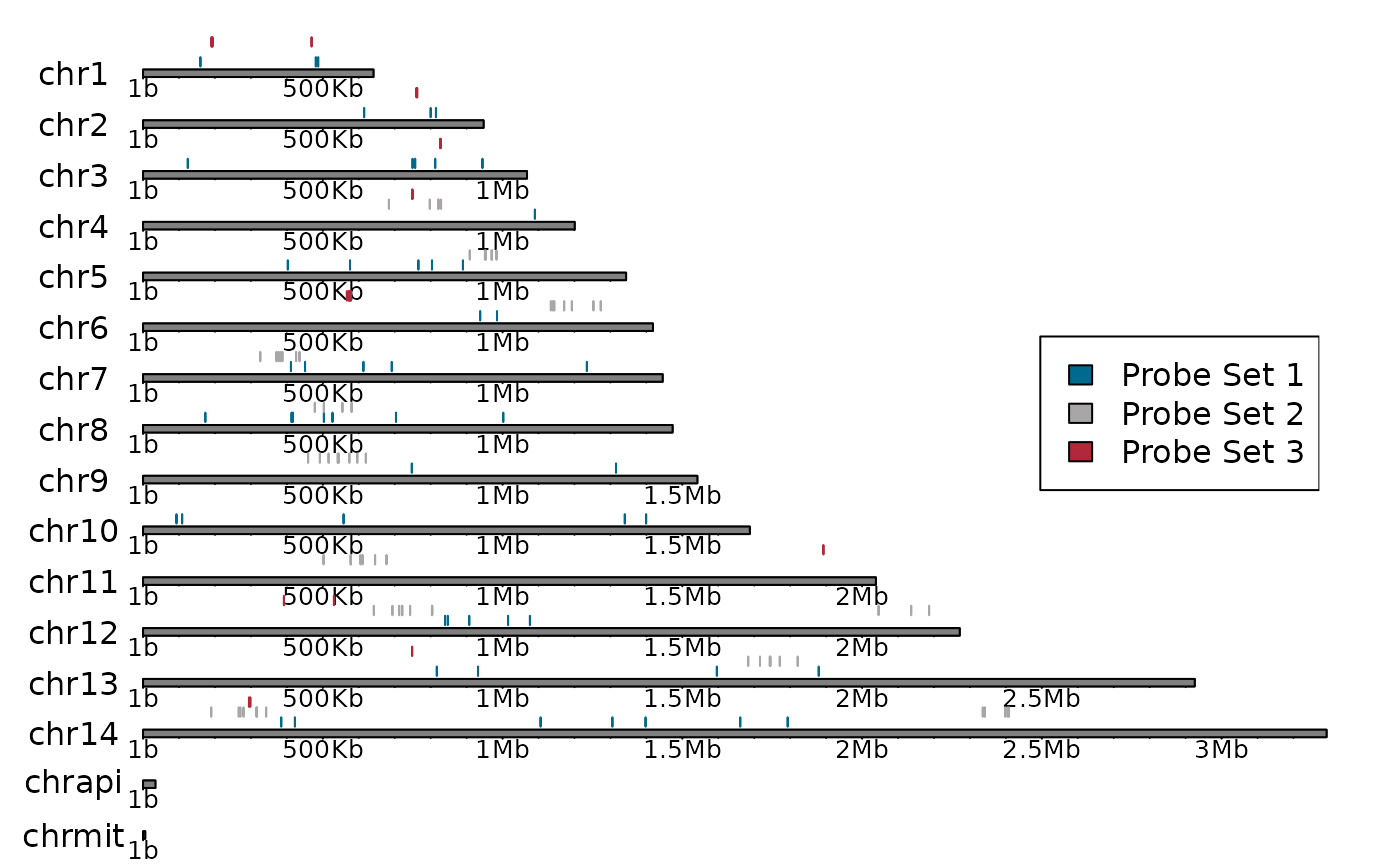
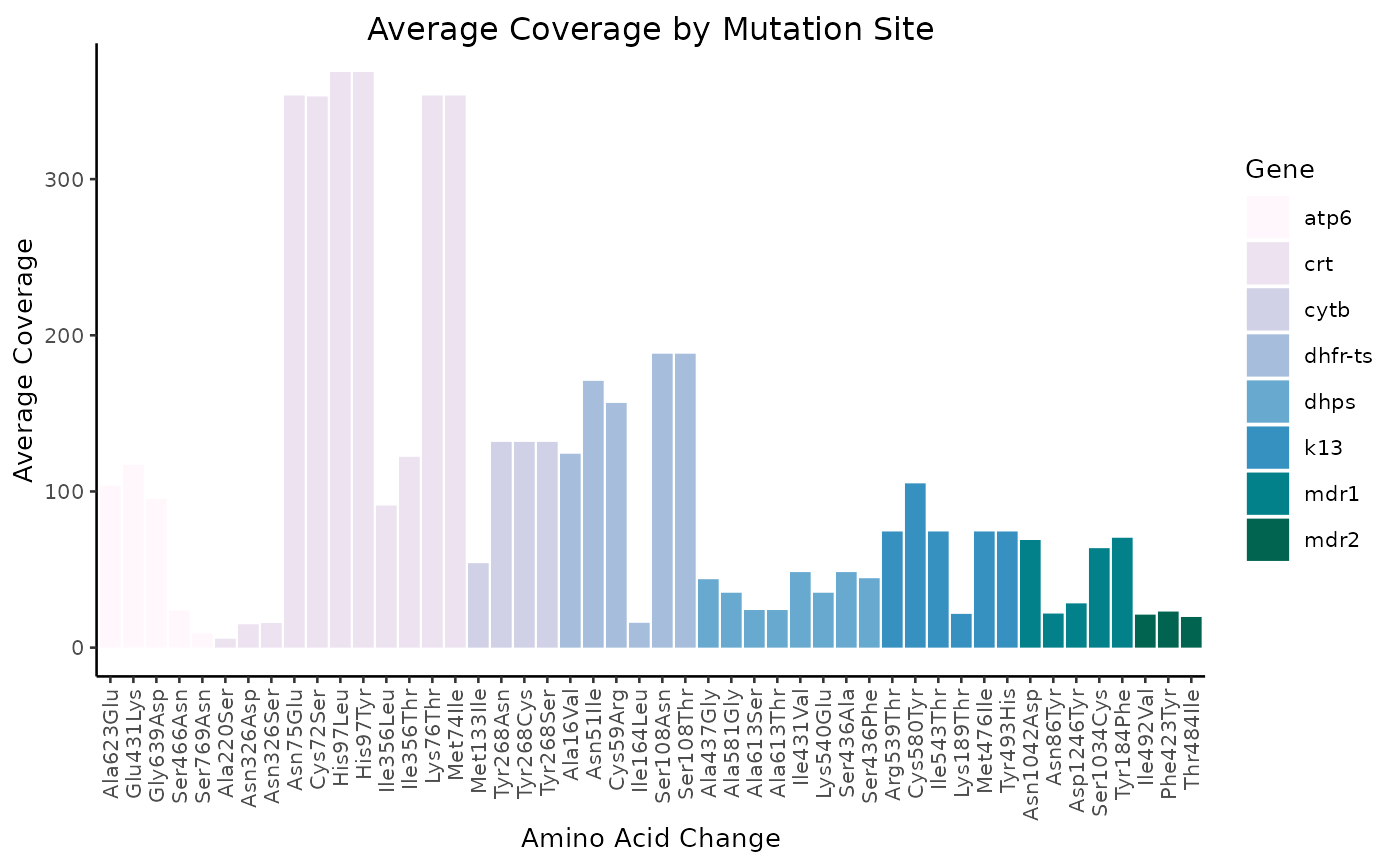
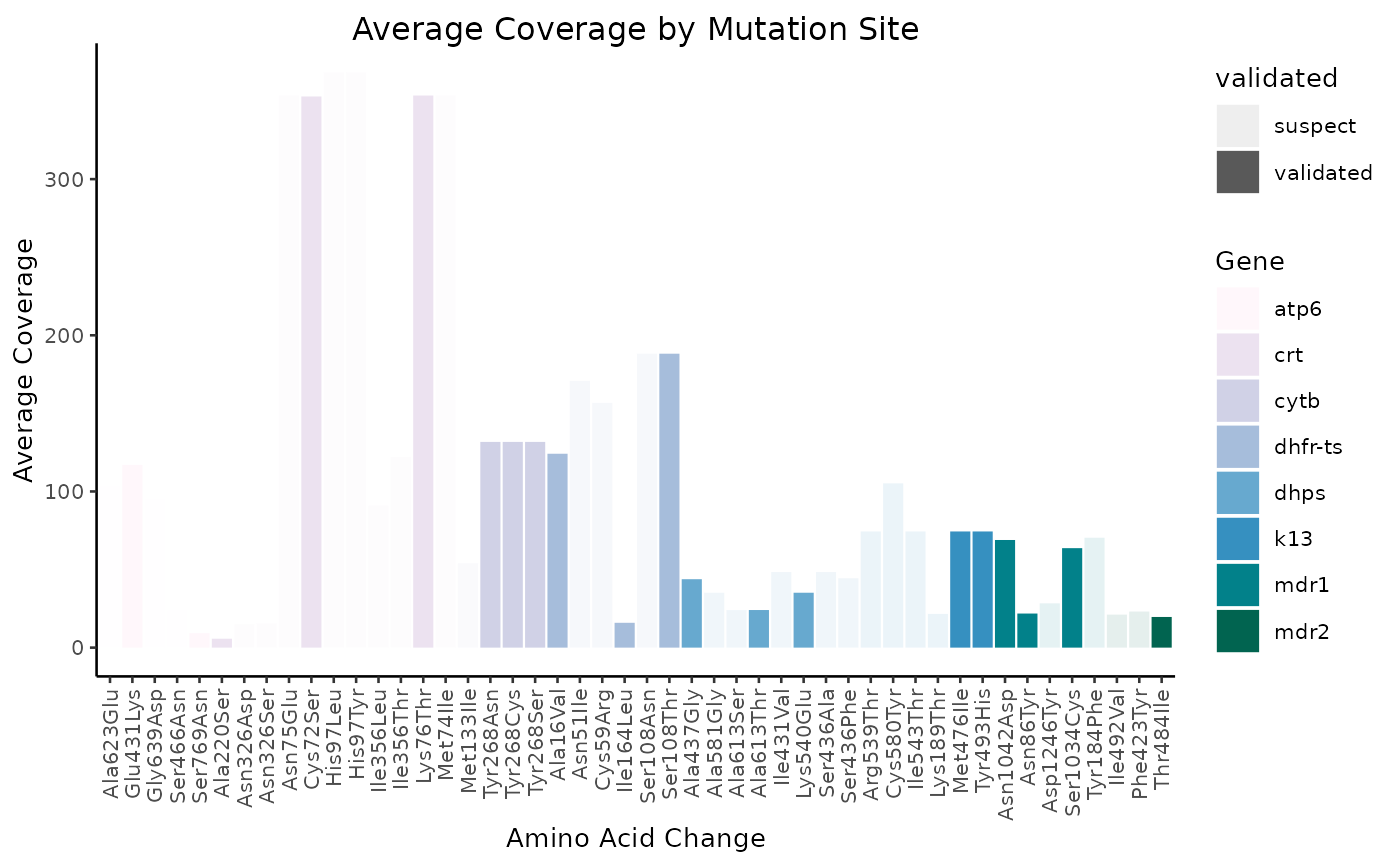
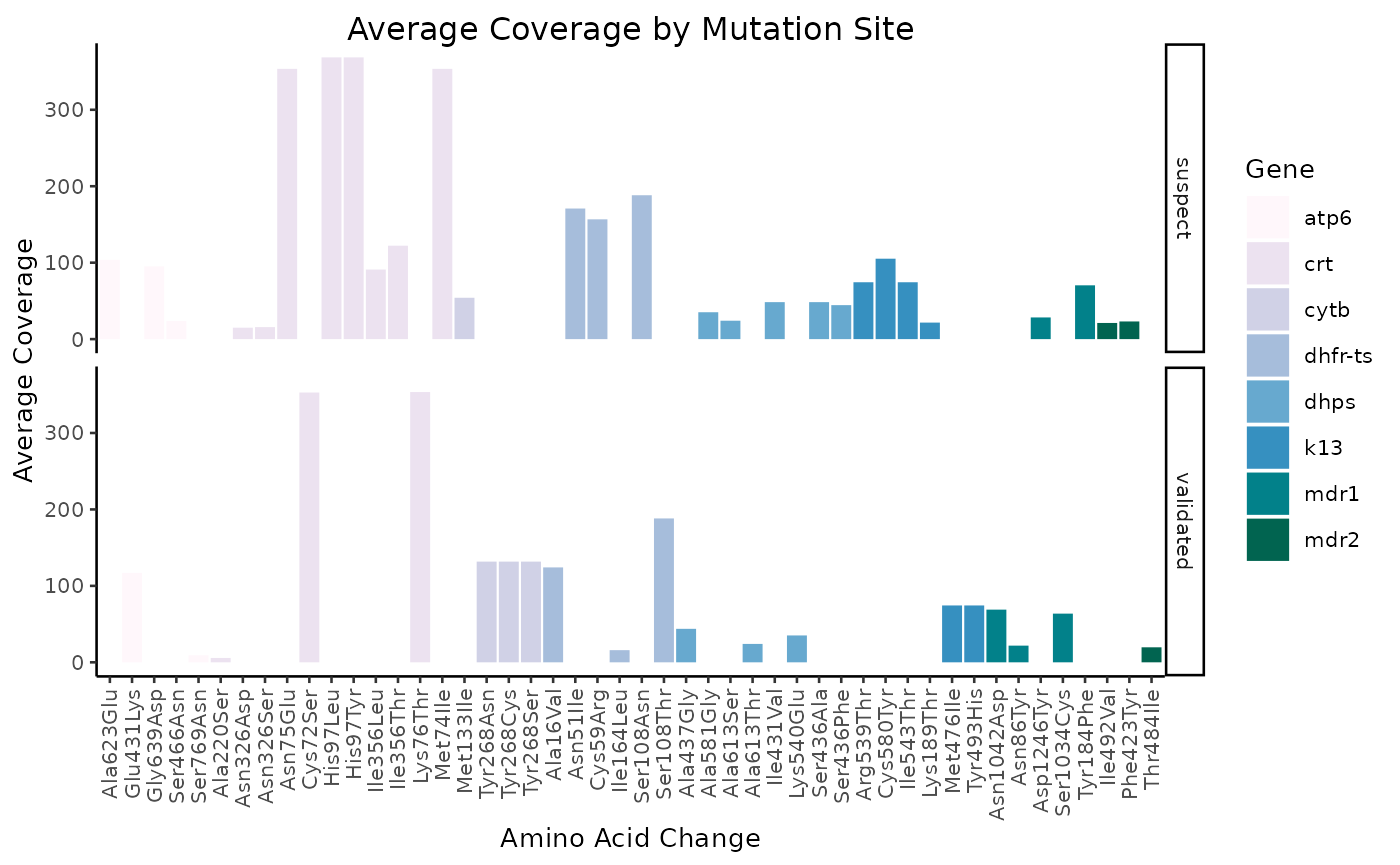
Average coverage
N.B. the remaining figures have not yet been incorporated into miplicorn, but as time goes on, more and more visualization methods will be added.